Understanding and exploiting druggable conformations
Using COVALfinder® in the early stages of drug discovery:
- Allows monitoring of the impact of chemical changes on inhibitor binding mode.
- Offers structural information in the absence of crystal structures.
- Enables a better design of drugs with high efficacy, selectivity or higher barrier to resistance development.
FGFR1 inhibitors stabilize different binding modes
Kinases switch between active and inactive states upon interaction with specific modulators. These conformational changes involve movements of two structural motifs:
- The DFG motif, which can adopt an open conformation (DGF-in) or a closed and inactive conformation (DGF-out) that occludes the access of the substrate sites.
- The αC-helix, which has to be positioned correctly for efficient catalysis (αC-in). C-helix rotation (αC-out) results in an inactive state of the kinase.
Fibroblast growth factor receptors are a family of tyrosine kinases implicated in several cancers. COVALfinder® allows study of the shift between FGFR1 conformations upon drug binding or covalent modification.
Fisogatinib
Irreversible, isoform-selective FGFR4 compound optimized for clinical use. FGFR4 bears a cysteine at position 552 that reacts with fisogatinib whereas FGFR1 lacks a comparable cysteine at this location.
- Our analysis supports that the lack of the reactive nucleophile in FGFR1 prevents covalent modification.
- FGFR1-fisogatinib complex formation follows a reversible one-step mechanism governed by a low binding affinity (885 nM) that correlates with the reported inhibitory potency (506 nM)1.
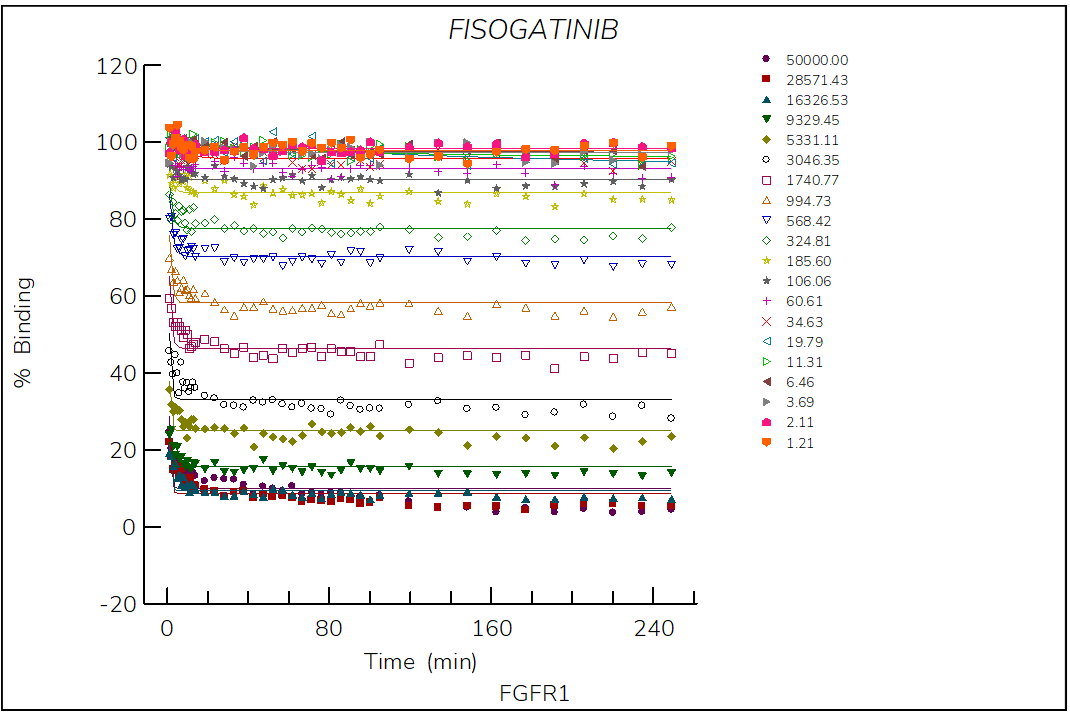
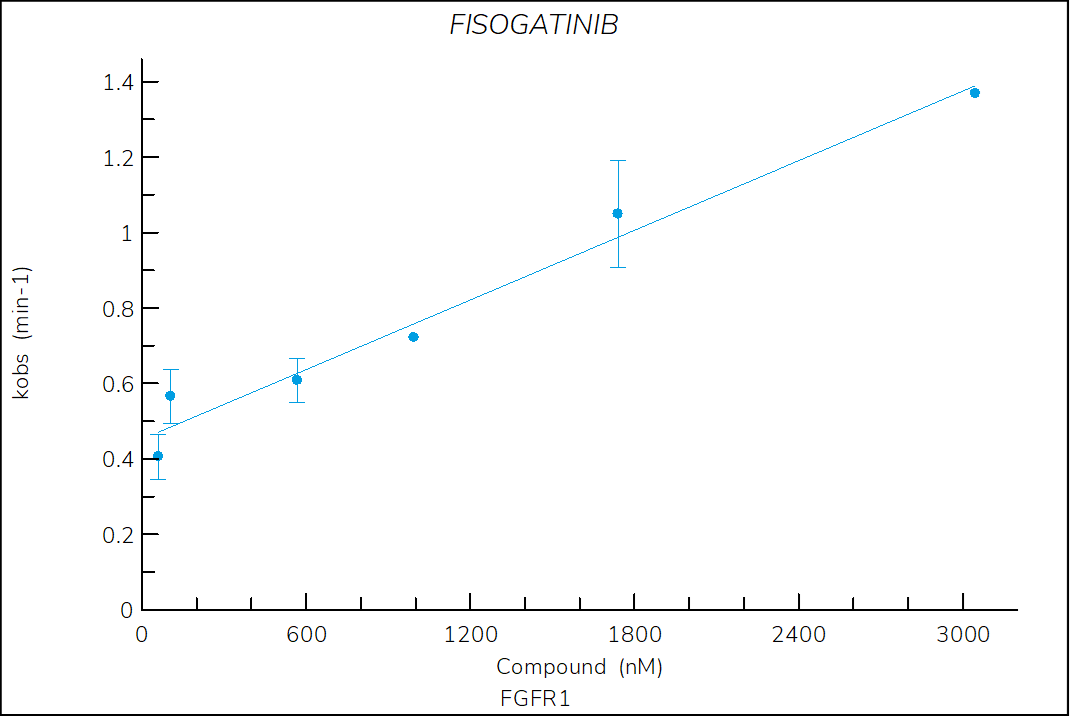

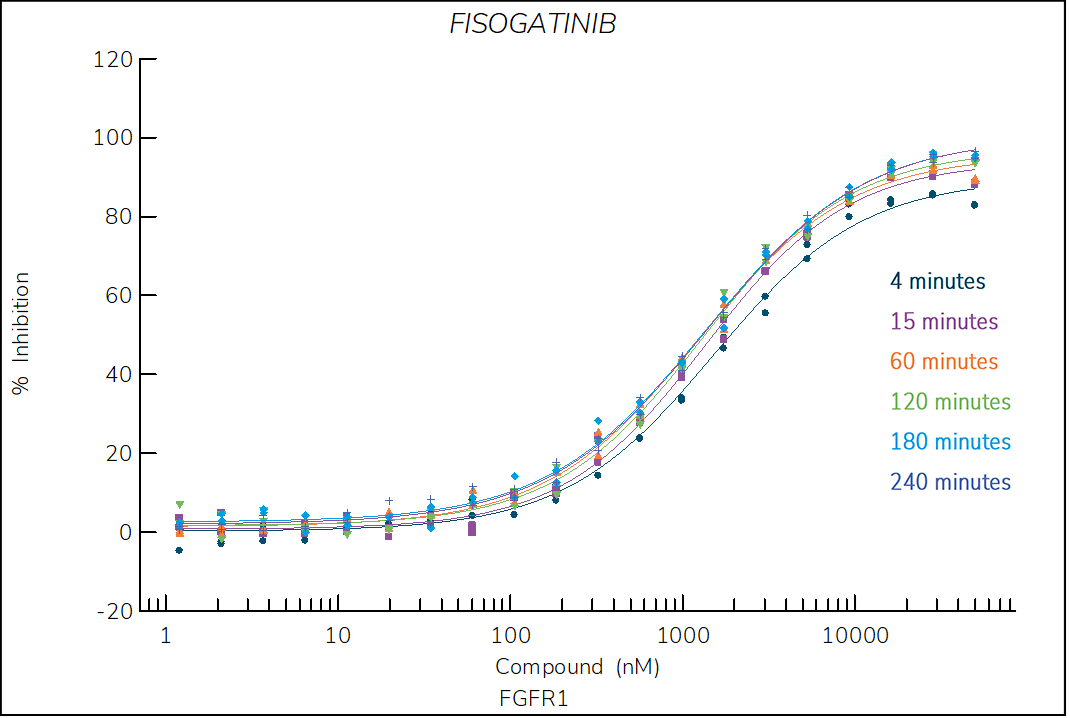
Figure 1. Progress curve of FGFR1 incubated with increasing compound concentrations, dependence of kobs on compound concentration, dose-response curves over time and IC50 values over time for fisogatinib. The complex formation follows a reversible one-step mechanism governed by the following constants: Kd: 885 nM, k1: 8.5×103 M-1s-1 and k2: 7.3×10-3 s‑1.
Ponatinib
Reversible, pan-FGFR FDA-approved drug classified as a type II inhibitor. The crystal structure of FGFR1 in complex with ponatinib shows that the inhibitor induces an inactive (αC-out) closed conformation (DFG-out)1.
- Our results confirm that FGFR1-ponatinib complex formation takes place in reversible sequential events.
- Ponatinib binding is the initial event and the displacement of the DGF motive and αC-helix the second, inducing the DFG-out, αC-out conformation. This is in line with previous studies2, 3.
- The high affinity of ponatinib (Kd*: 7.5 nM) is a result of the extraordinary stability of the FGFR1-ponatinib complex (koff: 1.4×10-4s-1). Both values correlate with those published in the literature2.

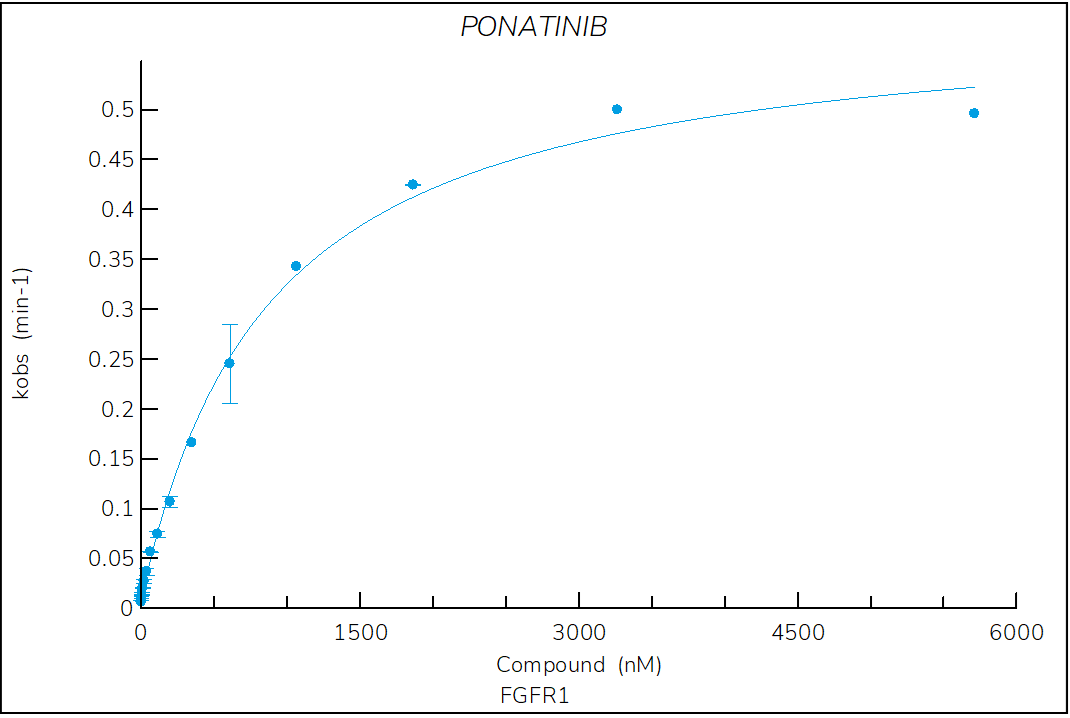


Figure 2. Progress curve of FGFR1 incubated with increasing compound concentrations, dependence of kobs on compound concentration, dose-response curves over time and IC50 values over time for ponatinib. The complex formation follows a reversible two-step mechanism governed by the following constants: Kd: 524 nM, Kd*: 7.5 nM, k3: 9.9×10-3 s-1 and k4: 1.4×10‑4 s‑1.
Erdafitinib
Reversible, pan-FGFR FDA-approved drug classified as a type I1/2 inhibitor. The structure of erdafitinib bound to FGFR1 shows that the compound binds to an inactive (αC-out) open (DFG-in) conformation of FGFR11.
- The two-step reversible kinetics confirm that erdafitinib displaces the αC-helix, inducing the αC-out conformation upon binding.
- The lower stability of the FGFR1-erdafitinib complex compared to ponatinib is consistent with the lack of a DFG-flip.
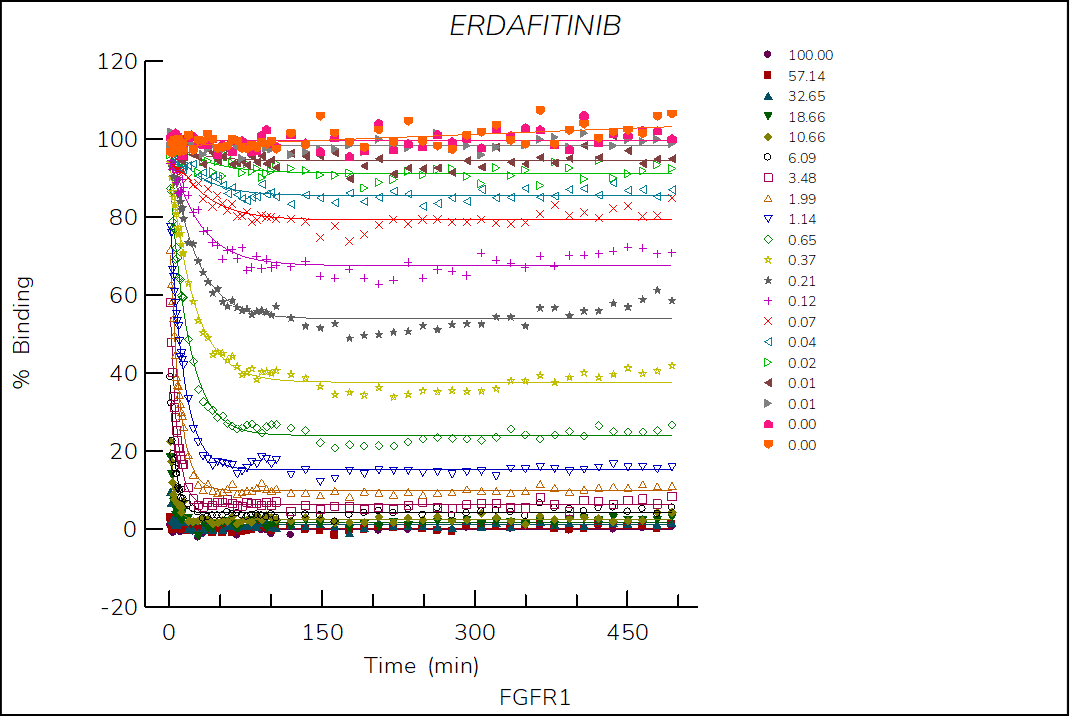
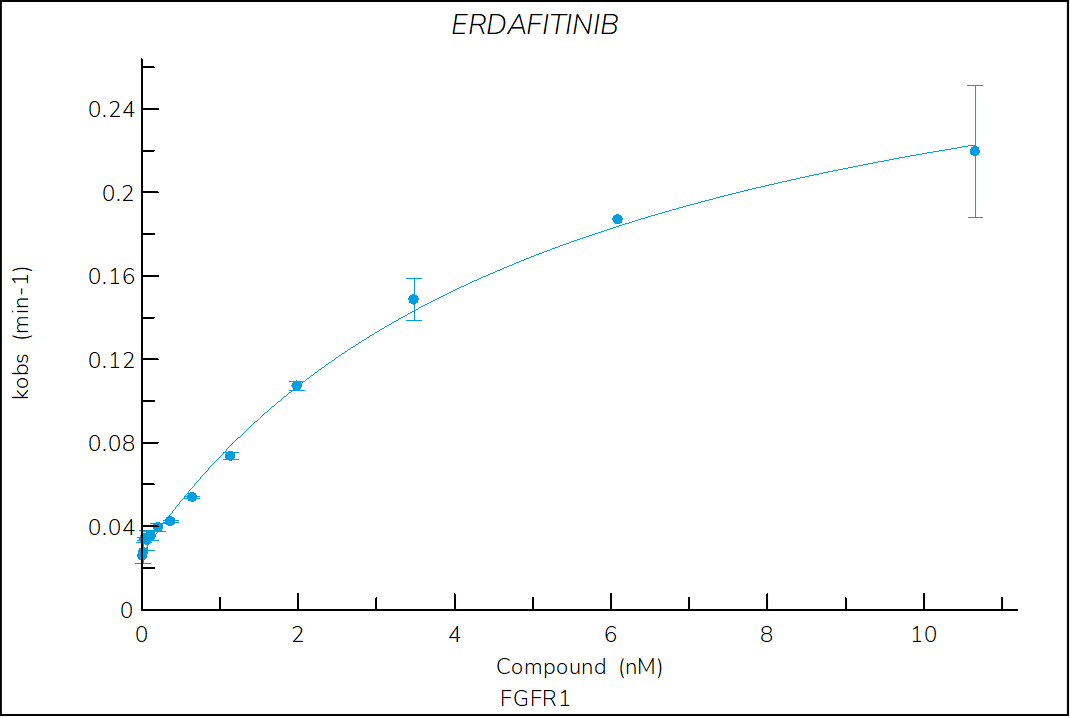
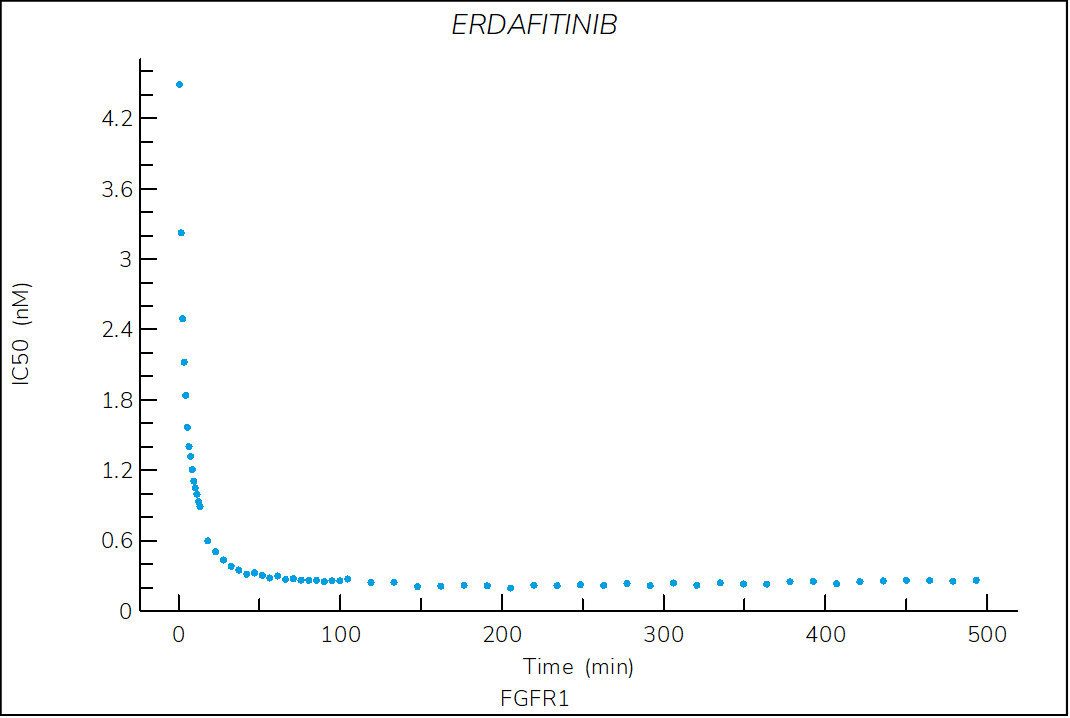

Figure 3. Progress curve of FGFR1 incubated with increasing compound concentrations, dependence of kobs on compound concentration, dose-response curves over time and IC50 values over time for erdafitinib. The complex formation follows a reversible two-step mechanism governed by the following constants: Kd: 3.2 nM, Kd*: 0.3 nM, k3: 4.9×10-3 s-1 and k4: 4.4×10-4 s‑1.
Futibatinib
Irreversible, pan-FGFR inhibitor optimized for clinical use. The structure of futibatinib bound to FGFR1 shows that the compound forms a covalent Michael adduct with cysteine 4881.
- The dependence of kobs on compound concentration confirm a two-step, time-dependent inhibition with a zero y-intercept, which is a hallmark of irreversible inhibitors.

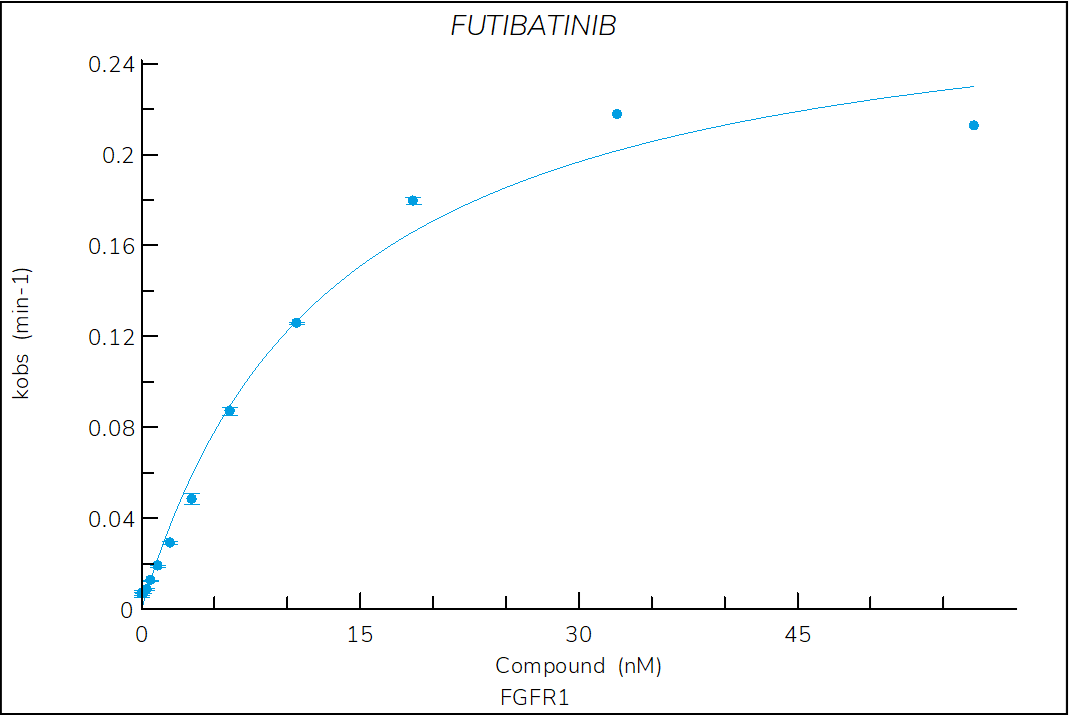

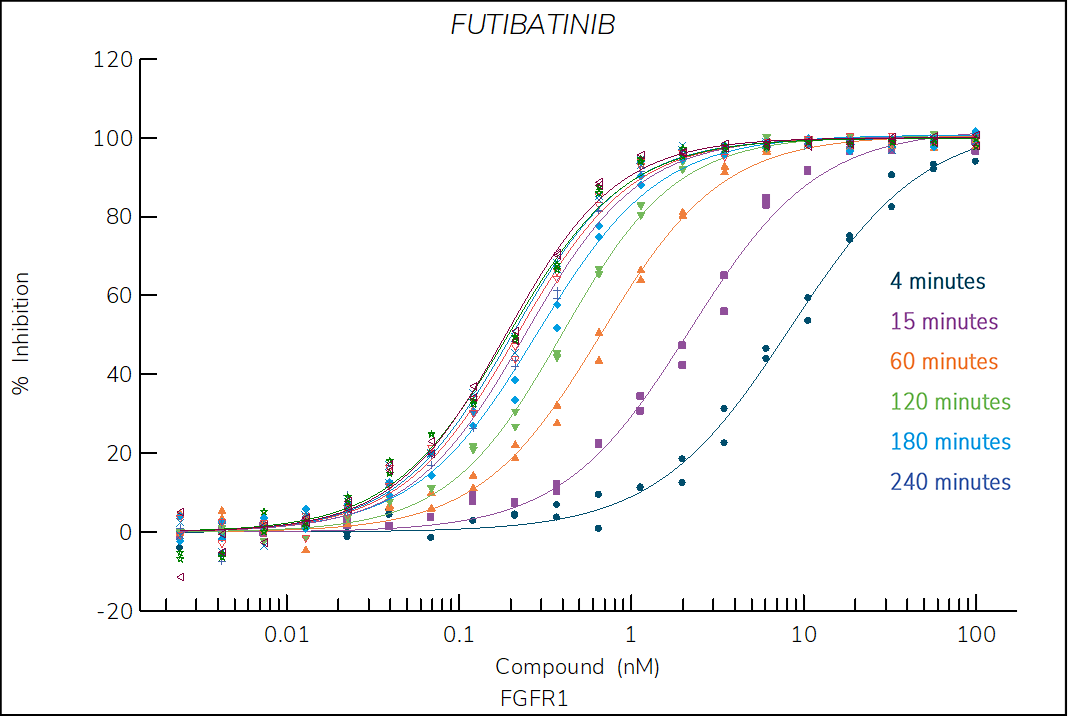
Figure 4. Progress curve of FGFR1 incubated with increasing compound concentrations, dependence of kobs on compound concentration, dose-response curves over time and IC50 values over time for futibatinib. The complex formation follows a irreversible two-step mechanism governed by the following constants: KI: 7.9 nM, kinact: 4.7×10-3 s-1 and kinact/KI: 6.0×105 M-1s‑1.
References
- Roskoski R. (2020) The role of fibroblast growth factor receptor (FGFR) protein-tyrosine kinase inhibitors in the treatment of cancers including those of the urinary bladder. Pharmacol Res 151:104567.
- Tucker J.A. et al. (2014) Structural insights into FGFR kinase isoform selectivity: diverse binding modes of AZD4547 and ponatinib in complex with FGFR1 and FGFR4. Structure. 2;22(12):1764-1774.
- Klein T. et al. (2015) Structural and dynamic insights into the energetics of activation loop rearrangement in FGFR1 kinase. Nat Commun. 6:7877.